当前计算凝聚态物理中QMC是非常常用的第一性原理计算方法之一(其他还包括密度泛函理论DFT等,之后肯定会写),当今QMC领域与前沿机器学习方法有许多交叉,不过这篇文章将主要以介绍传统概念为主。
Ⅰ. 第一性原理(Ab initio)
1. 多体薛定谔方程
计算凝聚态领域,第一性原理的源头自然就是多体薛定谔方程,一个由 $M$ 个原子核(每个原子核电荷量为 $Z_I e$,质量为 $M_I$)和 $N$ 个电子(每个电子电荷量为 $-e$,质量为 $m_e$)组成的系统,其完整的非相对论哈密顿量由 5 个部分组成:
\[\hat{H} = \hat{T}_n + \hat{T}_e + \hat{V}_{n-n} + \hat{V}_{e-e} + \hat{V}_{n-e}\]
具体数学形式如下:
\[\hat{H} = -\sum_{I=1}^{M} \frac{\hbar^2}{2M_I} \nabla_I^2 - \sum_{i=1}^{N} \frac{\hbar^2}{2m_e} \nabla_i^2 + \sum_{I < J}^{M} \frac{Z_I Z_J e^2}{4\pi\varepsilon_0 |\mathbf{R}_I - \mathbf{R}_J|} + \sum_{i < j}^{N} \frac{e^2}{4\pi\varepsilon_0 |\mathbf{r}_i - \mathbf{r}_j|} - \sum_{i=1}^{N}\sum_{I=1}^{M} \frac{Z_I e^2}{4\pi\varepsilon_0 |\mathbf{r}_i - \mathbf{R}_I|}\]
一般具体问题中我们都可以采Born-Oppenheimer近似,将原子核动能项 $\hat{T}n$ 设为 0,并将核-核排斥项 $\hat{V}{n-n}$ 视为一个常数,其电子哈密顿量简化为:
\[\hat{H}_e = -\sum_{i=1}^{N} \frac{\hbar^2}{2m_e} \nabla_i^2 + \sum_{i < j}^{N} \frac{e^2}{4\pi\varepsilon_0 |\mathbf{r}_i - \mathbf{r}_j|} + \sum_{i=1}^{N} v_{ext}(\mathbf{r}_i)\]
| 其中 $v_{ext}(\mathbf{r}i) = -\sum{I=1}^{M} \frac{Z_I e^2}{4\pi\varepsilon_0 |
\mathbf{r}_i - \mathbf{R}_I |
}$ 为电子感受到的外势场。 |
2. Hartree-Fock Method
哈特里-福克方法(HF 方法)是求解多体薛定谔方程最经典的第一性原理近似方法。为了解开多电子体系乱七八糟的薛定谔方程,做出了几个物理假设:
-
单粒子近似
整个体系的多体波函数,可以拆解为由一个个独立的单电子波函数(轨道 $\psi_i$)组合而成。
-
Slater行列式
多体波函数的形式为单个Slater行列式
\[\Psi(\mathbf{x}_1, \mathbf{x}_2, \dots, \mathbf{x}_N) = \frac{1}{\sqrt{N!}} \begin{vmatrix} \psi_1(\mathbf{x}_1) & \psi_2(\mathbf{x}_1) & \cdots & \psi_N(\mathbf{x}_1) \\ \psi_1(\mathbf{x}_2) & \psi_2(\mathbf{x}_2) & \cdots & \psi_N(\mathbf{x}_2) \\ \vdots & \vdots & \ddots & \vdots \\ \psi_1(\mathbf{x}_N) & \psi_2(\mathbf{x}_N) & \cdots & \psi_N(\mathbf{x}_N) \end{vmatrix}\]
由此可以写出能量表达式
\(E = \int \Psi^*(\mathbf{x}_1, \mathbf{x}_2,...,\mathbf{x}_N) \hat{H}_e \Psi(\mathbf{x}_1, \mathbf{x}_2,...,\mathbf{x}_N) d^N\mathbf{x}\)
对单个单电子波函数作变分
\(\frac{\delta E}{\delta \psi_i} = 0\)
可以得到单电子波函数的HF方程
\(\hat{f} \psi_i(\mathbf{x}) = \varepsilon_i \psi_i(\mathbf{x})\)
这里 $\varepsilon_i$ 是该轨道的单粒子能量,而 $\hat{f}$ 称为 Fock算符,其完整展开形式为:
\[\hat{f} = -\frac{\hbar^2}{2m_e}\nabla^2 + v_{ext}(\mathbf{r}) + \hat{J} - \hat{K}\]
其中前两项是传统单电子物理项,后两项分别体现了多电子的平均场近似和交换反对称性的等效排斥效应
平均场近似项
\[\hat{J}\psi_i(\mathbf{x}_1) = \left( \sum_{j=1}^{N} \int \frac{e^2 |\psi_j(\mathbf{x}_2)|^2}{4\pi\varepsilon_0 |\mathbf{r}_1 - \mathbf{r}_2|} d\mathbf{x}_2 \right) \psi_i(\mathbf{x}_1)\]
- 不难看出它把其他电子看作一团连续的静止电荷云,计算当前电子在这团电荷云中受到的排斥力。
交换项
\[\hat{K}\psi_i(\mathbf{x}_1) = \left( \sum_{j=1}^{N} \int \frac{e^2 \psi_j^*(\mathbf{x}_2) \psi_i(\mathbf{x}_2)}{4\pi\varepsilon_0 |\mathbf{r}_1 - \mathbf{r}_2|} d\mathbf{x}_2 \right) \psi_j(\mathbf{x}_1)\]
- 这一项在空间中拉开相同自旋电子之间的距离,可以说就是由于交换反对称性体现的等效排斥效应。
个人理解是,如果说薛定谔方程本身是在受限(交换反对称性限制)的希尔伯特空间寻找本征解,那么HF近似的本质,可以看作是在空间的一个子集(单行列式)里寻找最优解。这种方法确实可以找到一个基态的近似,但其他凑出来的非基态解就显得没什么用。
咳咳,好了以上其实和QMC没什么关系)))只是一点背景介绍,总之QMC的直接拟合目标就是多体系统的波函数$ \Psi $,而主要实现思路则有变分蒙特卡洛和扩散蒙特卡洛两种。
Ⅱ. 变分蒙特卡洛(VMC)
1. 基本原理
首先我们肯定知道,试探波函数 $\Psi(\mathbf{R}; \boldsymbol{\alpha})$(这里波函数并不一定归一化,因为高维空间求归一化因子本身很耗算力)它关于系统哈密顿量 $\hat{H}$ 的能量期望值 $E(\boldsymbol{\alpha})$,永远大于或等于系统的基态能量 $E_0$:
\[E(\boldsymbol{\alpha}) = \frac{\langle \Psi(\boldsymbol{\alpha}) | \hat{H} | \Psi(\boldsymbol{\alpha}) \rangle}{\langle \Psi(\boldsymbol{\alpha}) | \Psi(\boldsymbol{\alpha}) \rangle} \ge E_0\]
- $\boldsymbol{\alpha} = (\alpha_1, \alpha_2, \dots, \alpha_m)$ 是变分参数(例如轨道指数、Jastrow因子的参数,现代方法已经有模型开始使用神经网络权重)。
所以目标自然就是寻找一组最优参数 $\boldsymbol{\alpha}^*$,使得 $E(\boldsymbol{\alpha})$ 最小,这件事和神经网络梯度下降本身非常的相似。甚至损失函数就是能量本身,非常典型的无监督学习。
如果直接去写上述期望值的坐标表象积分,我们会得到:
\[E(\boldsymbol{\alpha}) = \frac{\int \Psi^*(\mathbf{R}; \boldsymbol{\alpha}) \hat{H} \Psi(\mathbf{R}; \boldsymbol{\alpha}) d\mathbf{R}}{\int |\Psi(\mathbf{R}; \boldsymbol{\alpha})|^2 d\mathbf{R}}\]
进行一个简单的代数变换
\[E(\boldsymbol{\alpha}) = \int \left( \frac{|\Psi(\mathbf{R}; \boldsymbol{\alpha})|^2}{\int |\Psi(\mathbf{R}'; \boldsymbol{\alpha})|^2 d\mathbf{R}'} \right) \cdot \left( \frac{\hat{H} \Psi(\mathbf{R}; \boldsymbol{\alpha})}{\Psi(\mathbf{R}; \boldsymbol{\alpha})} \right) d\mathbf{R}\]
现在,这个公式可以拆为两部分:
A. 概率密度函数 (PDF)
\[P(\mathbf{R}) = \frac{|\Psi(\mathbf{R}; \boldsymbol{\alpha})|^2}{\int |\Psi(\mathbf{R}'; \boldsymbol{\alpha})|^2 d\mathbf{R}'}\]
B. 局部能量 (Local Energy)
定义一个算符作用于波函数后的比值函数,称为局部能量 $E_L(\mathbf{R})$:
\[E_L(\mathbf{R}) = \frac{\hat{H} \Psi(\mathbf{R}; \boldsymbol{\alpha})}{\Psi(\mathbf{R}; \boldsymbol{\alpha})}\]
最终,我们的奖励函数,也就是能量期望值可以换个角度理解为局部能量在概率分布 $P(\mathbf{R})$ 下的期望:
\[E(\boldsymbol{\alpha}) = \int P(\mathbf{R}) E_L(\mathbf{R}) d\mathbf{R} = \langle E_L \rangle_P\]
2. 蒙特卡洛积分与马尔可夫链采样
有了上述期望值形式,利用蒙特卡洛方法,将高维积分转化为随机采样求和:
\[E(\boldsymbol{\alpha}) \approx \bar{E} = \frac{1}{M} \sum_{i=1}^M E_L(\mathbf{R}_i)\]
当采样数 $M \to \infty$ 时,统计平均值 $\bar{E}$ 趋近于真实的期望值 $E(\boldsymbol{\alpha})$。其统计误差(标准差)为 $\sigma / \sqrt{M}$。
由于我们不知道 $P(\mathbf{R})$ 分母上的归一化常数(即那个高维积分本身),可以采用Metropolis 算法来构造马尔可夫链,使得采样点walker的分布正好符合波函数概率密度的分布
\[A(\mathbf{R} \to \mathbf{R}') = \min\left(1, \frac{P(\mathbf{R}')}{P(\mathbf{R})}\right) = \min\left(1, \frac{|\Psi(\mathbf{R}'; \boldsymbol{\alpha})|^2}{|\Psi(\mathbf{R}; \boldsymbol{\alpha})|^2}\right)\]
| 即通过随机游走,计算游走前后的波函数概率相对值,若相对值大于1,则接受新状态 $\mathbf{R} \to \mathbf{R}’$;否则以概率$ \frac{ |
\Psi(\mathbf{R}’; \boldsymbol{\alpha}) |
^2}{ |
\Psi(\mathbf{R}; \boldsymbol{\alpha}) |
^2} $接受新状态$\mathbf{R} \to \mathbf{R}’$。 |
接下来的思路很简单,对能量进行参数空间${\alpha_i}$进行梯度下降即可,具体的方法则和拟设的波函数形式有关,而如何拟设恰恰是VMC最大的问题。
3. 如何拟设(ansatz)
不难发现,VMC理想很美好,但是最大的问题就是,其能达到的精度上限完全由拟设的质量决定,而我们根本没办法写出一个完美的拟设函数形式$\Psi(\mathbf{R}; \boldsymbol{\alpha})$,只能在一些经验凑出的参数空间里强行拟合。
神经网络这种靠暴力堆参数的我们先暂且不谈,来介绍几个相对合理且常用的拟设形式。
一个好的拟设起码需要满足三个基本要求:满足费米子反对称性、满足尖峰条件(Cusp Conditions)、并且能高效计算波函数的值及其一阶和二阶导数(用于动能计算)。
A. Slater-Jastrow Ansatz
这算是 VMC 中最经典、应用最广泛的拟设。它的数学核心是将描述单粒子行为的Slater行列式与描述电子间关联的Jastrow因子相乘。
\[\Psi_{\text{SJ}}(\mathbf{R}) = \mathcal{J}(\mathbf{R}) \sum_{m=1}^{N_{det}} c_m D_m^{\uparrow}(\mathbf{R}_{\uparrow}) D_m^{\downarrow}(\mathbf{R_\downarrow})\]
Slater Determinant一般是用多行列式的线性叠加,其内部的单电子波函数$ \psi_i $一般通常预先通过HF方程或密度泛函理论(DFT)计算得到,其基组可以是高斯基(分子系统)或平面波基(周期性固体)。
由于行列式忽略了电子间的即时排斥。我们通过Jastrow 因子用来强行引入电子关联。最常用的形式是:
\[\mathcal{J}(\mathbf{R}) = \exp \left( \sum_{i} \chi(\mathbf{r}_i) + \sum_{i<j} u(\mathbf{r}_{ij}) + \sum_{I, i<j} f_I(\mathbf{r}_{iI}, \mathbf{r}_{jI}, \mathbf{r}_{ij}) \right)\]
- 一体项 $\chi(\mathbf{r}_i)$:电子-原子核关联,用于微调原子附近的电子密度。
- **二体项 $u(\mathbf{r}{ij})$:电子-电子关联。它的数学形式(如 Pade 近似)被精心设计,用以显式满足尖峰条件**(当 $r{ij} \to 0$ 时消除库仑势能的发散)。
- 三体项 $f_I$:电子-电子-原子核三体关联,提供更高级的微调。
对应的变分参数通常是多项式展开的系数。
B. 强关联系统:Jastrow-Slater-Backflow ansatz
在电子密度很高或强关联的系统(如液氦-3、均匀电子气或超导材料)中,电子的运动会引起周围电子海的集体屏蔽,单个电子就像包裹着一层乌云(准粒子)。传统的固定轨道行列式无法很好地描述这种现象。回流拟设(Backflow Ansatz)可以一定程度解决这个问题,但由于自己也不太懂,这里就不乱写了。
Ⅲ. 扩散蒙特卡洛(DMC)
1.基本原理
相比VMC的简单粗暴,DMC的想法要天才得多,对于求解含时薛定谔方程(取$\hbar=1$ )
\(i\frac{\partial \Psi(\mathbf{R}, t)}{\partial t} = \hat{H}\Psi(\mathbf{R}, t)\)
把时间替换为虚时间 $\tau = it$,含时薛定谔方程变成
\[-\frac{\partial \Psi(\mathbf{R}, \tau)}{\partial \tau} = \left(\hat{H} - E_T\right)\Psi(\mathbf{R}, \tau)\]
其中 $E_T$ 是一个常数偏移(试探能量值,相当于选取一个基态能量,对原薛定谔方程本身没什么影响)。则理论上通解可以写作
\[\Psi(\mathbf{R}, \tau) = e^{-(\hat{H} - E_T)\tau}\,\Psi(\mathbf{R}, 0)
= \sum_n c_n\,e^{-(E_n - E_T)\tau}\,\psi_n(\mathbf{R}).\]
这就很有意思了,因为可以发现虚时间演化本质上是一个过滤器。如果选取$E_T=E_0$,当 $\tau \to \infty$,所有激发态($E_n > E_0$)相对基态以 $e^{-(E_n - E_0)\tau}$ 的速率指数衰减;只要初态与基态不正交($c_0 \neq 0$),剩下的就只有基态分量$\psi_0$。用蒙特卡洛的视角,似乎只要让一堆随机选取的walker采样点在这个方程下自由演化,就能得到我们需要的基态波函数了。(当然肯定是不可能让你捡这么大便宜的,还是要对NP-hard有基本的尊重)
具体一点,现在的演化方程形式为
\[\frac{\partial \Psi(\mathbf{R}, \tau)}{\partial \tau}
= \underbrace{\frac{1}{2}\nabla^2 \Psi}_{\text{扩散项}}
- \underbrace{\bigl[V(\mathbf{R}) - E_T\bigr]\Psi}_{\text{分支项}}.\]
形式上就是一个自带源/汇的扩散方程
| 数学项 |
随机过程对应 |
| $\tfrac{1}{2}\nabla^2 \Psi$ |
扩散:高斯随机游走,扩散系数 $D = 1/2$ |
| $-(V - E_T)\Psi$ |
分支:walker 按 $V(x) > E_T$ 处死亡、$V(x) < E_T$ 处复制 |
注意 $\Psi$ 必须是正的才能把walker的分布解释为波函数分布。对玻色子基态这天然成立(基态无节点),对费米子就显然不可能了,这就是DMC最致命的符号问题,绕不开的大坑,后面再讲。
2.演化过程
精确的演化算子 $e^{-(\hat{H} - E_T)\Delta\tau}$ 没法直接计算,但当选取小步长$\Delta\tau$ ,可以用 Trotter 分解(一阶):
\[e^{-(\hat{T} + \hat{V} - E_T)\Delta\tau}
\approx e^{-\hat{T}\Delta\tau}\, e^{-(\hat{V} - E_T)\Delta\tau} + \mathcal{O}(\Delta\tau^2).\]
两个因子各自有具体的物理意义,对应的格林函数是:
扩散部分
\[G_{\text{diff}}(x \to x'; \Delta\tau) = \frac{1}{\sqrt{2\pi\Delta\tau}}\,\exp\!\left[-\frac{(x'-x)^2}{2\Delta\tau}\right].\]
也就是步长服从 $\mathcal{N}(0, \sqrt{\Delta\tau})$ 的高斯随机游走。
分支部分
\[w= G_{\text{branch}}(x; \Delta\tau) = \exp\!\bigl[-(V(x) - E_T)\,\Delta\tau\bigr].\]
权重$w$的具体含义就是此处的walker密度流需要以权重$w$进行膨胀或收缩,每个 walker 携带这个权重,为了避免权重方差爆炸,把它转换成离散的复制数:
\[m = \lfloor w + u \rfloor,\quad u \sim \mathcal{U}(0, 1).\]
- $m = 0$:walker 消亡;
- $m = 1$:保留;
- $m \geq 2$:分裂为 $m$ 份。
期望复制数 $\langle m \rangle = w$,与连续权重一致。
如果 $E_T = E_0$,walker 总数的期望随时间不变;偏离时 walker 数会指数爆炸或灭绝。实际算法用反馈机制把总数稳定在目标值 $N_{\text{target}}$:
\[E_T \leftarrow \langle V \rangle_{\text{walkers}} \;-\; \alpha\,\ln\!\left(\frac{N}{N_{\text{target}}}\right),\]
其中 $\alpha$ 是自定义的反馈强度。如果$N$能保持稳定,理论上收敛后 $E_T$ 的时间平均就是基态能量 $E_0$ 的估计。
3.负符号问题和固定节点近似
不难注意到两个小问题,一是DMC的数学讨论似乎都没有设计到交换反对称性。这意味着如果真让它自然演化,最终剩下来的只会是能量更低的玻色子基态而不是我们需要的费米子基态。
另一个则是前面提到的符号问题,看起来,似乎简单地给walker带一个“+1”或者“-1”的符号标签就可以解决问题。确实如此,但当我们最后计算某个物理量 $O$ 的期望值时,我们需要做如下统计:\(\langle \hat{O} \rangle = \frac{\int \Psi^+ \hat{O} d\mathbf{R} - \int \Psi^- \hat{O} d\mathbf{R}}{\int \Psi^+ d\mathbf{R} - \int \Psi^- d\mathbf{R}} = \frac{\langle \hat{O} \rangle_+ - \langle \hat{O} \rangle_- \cdot z}{1 - z}\)
其中 $z = \frac{\int \Psi^- d\mathbf{R}}{\int \Psi^+ d\mathbf{R}}$ 是正负符号的重叠度。而费米子独特的交换反对称告诉我们其波函数正负符号的walker数期望值一定是完全相同的,这意味着分母的$1-z$会相当的小,由此得到的期望值信噪比将会非常高,几乎没法得到一个合理的统计值。
这两个问题的本质其实是一样的,因为费米子的基态波函数独特的交换反对称必然导致有多个区域是完全等价的,因此全局的统计完全可以转化为对单个区域的统计,整个空间的波函数可以通过这单个“格子”扩展出来。同时这样拓展出的波函数也能自然满足交换反对称。
于是所有问题的核心就是如何找到不同区域间的节点面,显然节点面为基态的零点,满足方程
\[\Psi_0(\mathbf{R})=0\]
一般的做法是利用VMC或者DFT求出一个试探解,把试探解的节点面当作真正节点面进行DMC演化得到最终解。如果节点面完全准确,那么理论上演化得到的答案是没有误差的,因此可以认为DMC的误差完全来源于节点面选取的精确程度。
Ⅳ. 实例:QMC求解均匀电子气模型
作为QMC最经典的工作之一,让我们来欣赏一下Cepeley发表在1980年PRL的代表性文章《Ground State of the Electron Gas by a Stochastic Method》
1.均匀电子气模型
先简单做一些背景介绍,在真实的金属中电子不仅受到相互之间的库仑排斥,还受到周期性排列、具有吸引力的原子核的拉扯。这种非均匀的环境让多体薛定谔方程几乎很难求解。
均匀电子气模型(jellium)把真实金属中的原子核简化为一团均匀分布的正电荷背景,只保留电子之间的库仑相互作用和电子的量子动能。
具体地说,把 $N$ 个点电荷电子放在体积 $V$ 中,再叠加一团均匀正电荷背景 $n_+(\mathbf r) = n = N/V$ 保证全局电中性。Hartree 原子单位($e = m_e = \hbar = 1$)下:
\[\hat H \;=\; \hat T \;+\; \hat V_{ee} \;+\; \hat V_{eb} \;+\; \hat V_{bb}\]
四项分别是:
| 项 |
表达式 |
物理含义 |
| 动能 |
$\hat T = -\dfrac{1}{2}\sum_{i=1}^N \nabla_i^{2}$ |
电子量子动能 |
| 电-电 |
$\hat V_{ee} = \dfrac{1}{2}\sum_{i\ne j}\dfrac{1}{\lvert\mathbf r_i - \mathbf r_j\rvert}$ |
电子两两排斥 |
| 电-背景 |
$\hat V_{eb} = -\sum_i \displaystyle\int_V d^3r’\,\dfrac{n}{\lvert\mathbf r_i - \mathbf r’\rvert}$ |
每个电子被背景吸引 |
| 背景-背景 |
$\hat V_{bb} = \dfrac{1}{2}\displaystyle\int_V d^3r\,d^3r’\,\dfrac{n^2}{\lvert\mathbf r - \mathbf r’\rvert}$ |
背景自能 |
如果$n$为常数,后面三个势能项在 $V\to\infty$ 时都是发散的,但数学上可以证明三项的和是收敛的。
\[\lim_{V\to\infty}\bigl(\hat V_{ee} + \hat V_{eb} + \hat V_{bb}\bigr) \;=\; \text{有限}\]
但具体数值计算我们不能硬着头皮用这个式子,在数值计算中,我们一般也不会在全空间进行采样,而是在 $L^3$ 立方盒子里用 周期边界条件 模拟无限大。此时的总静电能(对所有盒子求和)是
\[V_{\text{tot}} \;=\; \frac{1}{2}\sum_{i,j}\sum_{\mathbf n}{}'\,\frac{1}{\lvert\mathbf r_i - \mathbf r_j + \mathbf n L\rvert} \;-\; N\!\sum_{\mathbf n}\!\int_{V_{\text{cell}}}\!\frac{n}{\lvert\mathbf r - \mathbf r'\rvert}d^3r' \;+\; \frac{N^2}{2}\sum_{\mathbf n}\!\iint\!\frac{n^2}{\lvert\mathbf r - \mathbf r'\rvert}d^3r\,d^3r'\]
撇号意思是排除自相互作用 ${i=j, \mathbf n = 0}$。
数学上一通推导可以得到下面这个收敛的哈密顿量形式
\[\boxed{\;\hat H \;=\; -\frac{1}{2}\sum_i \nabla_i^{\,2} \;+\; \frac{1}{2}\sum_{i\ne j} v_E(\mathbf r_i - \mathbf r_j) \;+\; \frac{N}{2}\,v_M\;}\]
所有单粒子-单粒子(包括所有镜像、背景一切)的有效相互作用合并为Ewald 对势 $v_E(\mathbf r)$
\[{\;v_E(\mathbf r) \;=\; \sum_{\mathbf n}\frac{\operatorname{erfc}(\alpha\lvert\mathbf r + \mathbf n L\rvert)}{\lvert\mathbf r + \mathbf n L\rvert} \;+\; \frac{4\pi}{V}\sum_{\mathbf k\ne 0}\frac{e^{-k^2/4\alpha^2}}{k^2}\cos(\mathbf k\!\cdot\!\mathbf r)\;}\]
电子与自身镜像($i=j$ 但 $\mathbf n\ne 0$)的作用 + 与背景的零模贡献,记作Madelung 自像常数 $v_M$,对简单立方 Bravais 格子,这个数值化解出
\[v_M \;=\; -\xi_{sc}/L, \qquad \xi_{sc} = 2.8372974795\ldots\]
有了这个形式后,我们不难对模型进行一些定性的分析。选取一个无量纲数$r_s$来刻画电子密度
\[r_s \;=\; \left(\frac{3}{4\pi n}\right)^{1/3}, \qquad n = \frac{N}{L^3}\]
它就是每个电子占据体积的等效 Wigner-Seitz 半径,单位是 Bohr。
如果取两个极限,会发现均匀电子气存在相变现象
$r_s$ 极小:电子极密,相互拥挤,根据海森堡不确定性原理,电子动能巨大(动能 $\propto 1/r_s^2$),相互作用可以忽略不计,方程退化为自由费米子理想方程,其数学解为平面波行列式。
$r_s$ 极大:电子极稀,动能衰减极快,电子间的即时库仑排斥能(势能 $\propto 1/r_s$)主导系统,其数学解为空间离散位置的 $\delta$ 函数(即Wigner晶体相)
而二者间具体发生了什么事情,就需要我们的QMC来帮忙一探究竟了。
2.改进的DMC算法
相比起前面的DMC一般方程,论文中使用了重要性采样(Importance Sampling)方法
。引入试探波函数 $\Psi_T$ 引导采样,令 $f(\mathbf{R}, \tau) = \Psi_T(\mathbf{R})\Psi(\mathbf{R}, \tau)$,对原DMC演化方程两边同乘 $\Psi_T$,可以把方程变形为如下形式
\[\frac{\partial f}{\partial \tau} = \frac{1}{2}\nabla^2 f - \nabla \cdot \big[\mathbf{V}_D(\mathbf{R}) f\big] - \big[E_L(\mathbf{R}) - E_T\big] f\]
改进后的方程引入了漂移项($-\nabla \cdot [\mathbf{V}_D f]$),等效为引入了一个由 $\Psi_T$ 漂移力
\[\mathbf{V}_D(\mathbf{R}) = \nabla \ln |\Psi_T(\mathbf{R})|^2 = \frac{2\nabla \Psi_T(\mathbf{R})}{\Psi_T(\mathbf{R})}\]
直观理解,这个力可以把 Walker 从节面($\Psi_T=0$)处推开,并引导它们高效走向高概率区。也就是说$\Psi_T(\mathbf{R})$的形式其实直接决定了最重要的固定节点面($\Psi_T(\mathbf{R})=0$),同时合适的试探解$\Psi_T(\mathbf{R})$能够加速算法的收敛速度。
3. 拟设形式与计算结论
论文中试探解$\Psi_T(\mathbf{R})$来自VMC的初步迭代,采用的是经典的Slater-Jastrow Ansatz
\[\Psi_{\text{SJ}}(\mathbf{R}) = \mathcal{J}(\mathbf{R}) \sum_{m=1}^{N_{det}} c_m D_m^{\uparrow}(\mathbf{R}_{\uparrow}) D_m^{\downarrow}(\mathbf{R_\downarrow})\]
理论上随着$r_s$的变化,均匀电子气会经历三个相变过程

Jastrow因子在不同的相中大同小异,Slater多项式里的单电子波函数的形式才是相变的根本差异。为了拟合出论文中不同相的基态能量(顺磁费米流体 $E_{\text{PMF}}$、铁磁费米流体 $E_{\text{FMF}}$、维格纳晶体 $E_{\text{bcc}}$、以及用于对比的玻色流体 $E_{\text{BF}}$),必须为每一个相量身定制不同的试探波函数 $\Psi_T(\mathbf{R})$。
在戴维·塞珀利(David Ceperley)1980年的那篇划时代论文中,为了分别计算出四个不同的相(顺磁费米流体 $E_{\text{PMF}}$、铁磁费米流体 $E_{\text{FMF}}$、玻色流体 $E_{\text{BF}}$、以及维格纳晶体 $E_{\text{bcc}}$)的基态能量,他必须为每一个相量身定制不同的试探波函数 $\Psi_T(\mathbf{R})$。
顺磁费米流体($\Psi_T^{\text{PMF}}$):
动能主导,波函数由两个独立的Slater行列式相乘组成
\[\Phi_{\text{PMF}} = \det[\mathbf{D}_{\uparrow}] \times \det[\mathbf{D}_{\downarrow}]\]
矩阵元素为自由费米子的平面波解 $\exp(i \mathbf{k} \cdot \mathbf{r})$。
铁磁费米流体($\Psi_T^{\text{FMF}}$):
随着$r_s$增大,势能开始作为主导。电子为了减少在空间中相遇的概率慢慢趋于自旋相同,因此其波函数形式由一个单Slater行列式组成
\[\Phi_{\text{FMF}} = \det[\mathbf{D}_{\text{all}\uparrow}]\]
矩阵元素依然选取平面波 $\exp(i \mathbf{k} \cdot \mathbf{r})$。
玻色流体($\Psi_T^{\text{BF}}$):
作为其他相的对照组,整个试探波函数退化为纯粹的 Jastrow 形式
\[\Psi_T^{\text{BF}} = \prod_{i<j} e^{-u(r_{ij})}\]
维格纳晶体($\Psi_T^{\text{bcc}}$):
势能完全主导,单电子波函数必须体现出空间局域化,因此改用高斯型局域波函数作为骨架
\[\phi_{\mathbf{R}_I}(\mathbf{r}_i) = \exp\left( -C \|\mathbf{r}_i - \mathbf{R}_I\|^2 \right)\]
论文中对四种不同的波函数拟设在不同的$r_s$下进行VMC+DMC演化,得到了基态能量的分布$E_0(r_s)$
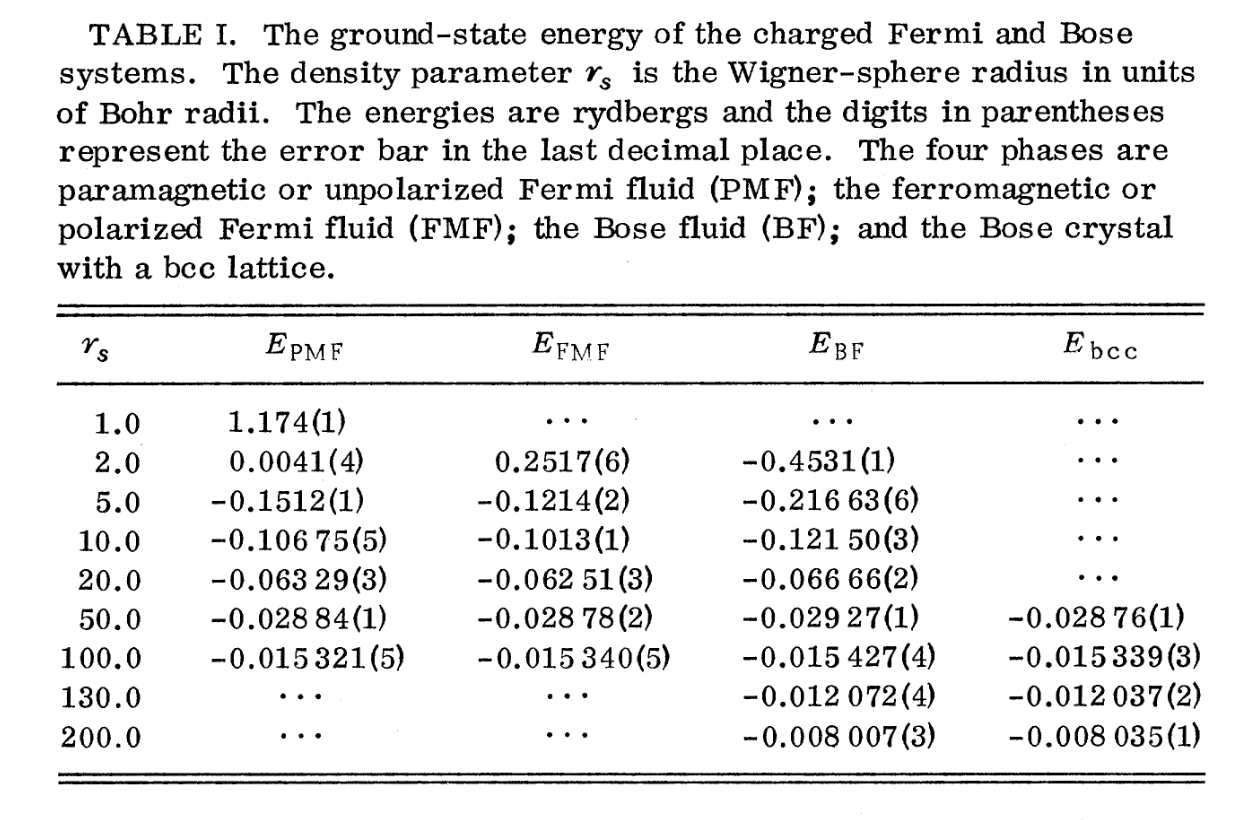
如果认为相同$r_s$下能量最低的为真实物态,则不难绘图看出整个模型的相变过程
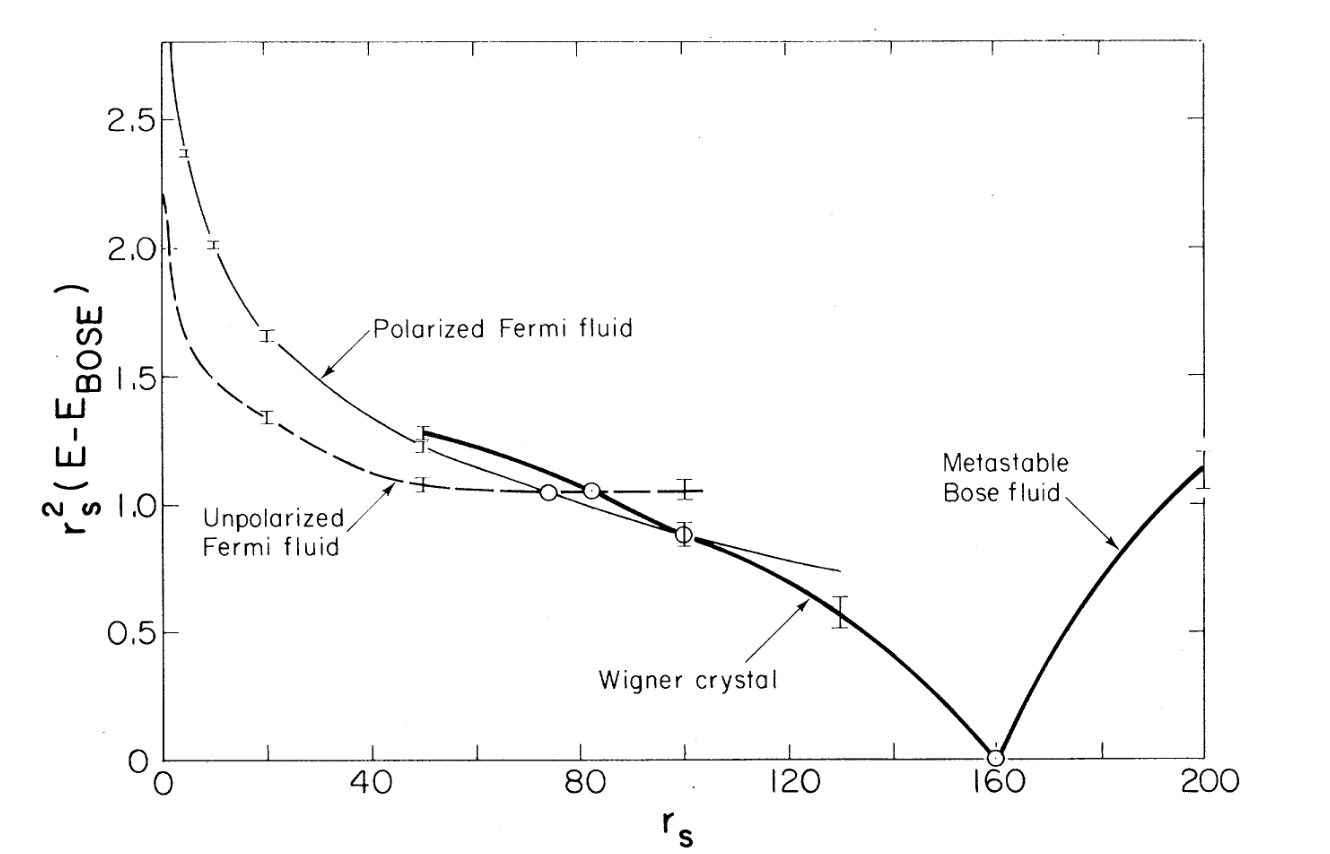
可以看见均匀电子气确实经历了从顺磁性费米流体到铁磁性费米流体到Wigner晶体的两次相变,与理论预测恰好一致,同时也反过来说明我们拟设的函数形式是合理的。
谨以此篇粗糙肤浅的文章代表自己科研工作的正式开始。
以上。
]]>