
密度泛函理论(DFT)应该算应用最广的量子力学数值计算方法了,在计算规模和精度上取得了不错的平衡(不像QMC大多数时候都只能算几十个原子),是学习计算凝聚态绕不过的一关。
60年前的理论应该还是很好看懂的吧,大概( 。
1. Hohenberg-Kohn 定理
1.1 从波函数到电荷密度
量子多体的第一性原理是多体薛定谔方程,在Born-Oppenheimer近似下,一个包含 $N$ 个电子的非相对论的哈密顿量可写为:
\[\hat{H} = \hat{T} + \hat{V}_{ee} + \hat{V}_{ext}\]具体展开长这样
\[\hat{H}_e = -\frac{1}{2} \sum_{i=1}^{N}\nabla_i^2 + \sum_{i < j}^{N} \frac{1}{|\mathbf{r}_i - \mathbf{r}_j|} + \sum_{i=1}^{N} v_{ext}(\mathbf{r}_i)\](因为懒所以)本文使用原子单位制($\hbar = m_e = e = 1$)
注意到,如果一个系统有 $N$ 个电子,它的波函数 $\Psi(x_1, x_2, …, x_N)$ [1] 就有 $3N$ 个空间自由度。当电子数量增加时,计算量指数级爆炸。也就是大家喜闻乐见(并非)的指数墙(Exponential Wall)。
DFT的天才设想就是将这个$3N$维多电子波函数转化为$3$维的电荷密度 $n(r)$
\[n(r) = N \sum_{s_1} \int \dots \int |\Psi(r, s_1, x_2, x_3, \dots, x_N)|^2 dx_2 dx_3 \dots dx_N\]不难看出这里的积分其实是全同性的体现,选择哪个变量做主元并不会有差别,前面乘以系数 $N$ 就行。
那么关键的问题其实是,这个看起来是从波函数中简化过来的东西,能不能和波函数一样告诉我们体系的物理量。这个想法听着就很大胆,但神奇的就是,系统的所有基态性质还真就是基态电子密度 $n(r)$ 的唯一泛函。
下面就来介绍一下DFT的基石,在1964年提出的 Hohenberg-Kohn 定理 [2]
1.2 HK 第一定理(唯一性定理)
Theorem 1:外势 $V_{ext}(r)$ 是基态电子密度 $n(r)$ 的唯一泛函(在相差一个常数项的意义下)。
唯一性定理大都异曲同工,先反证法起手。我们假设定理不成立。即假设存在两个本质上不同的外势 $V_{ext}^{(1)}(r)$ 和 $V_{ext}^{(2)}(r)$(即$V_{ext}^{(1)} - V_{ext}^{(2)} \neq \text{const}$),但它们经过薛定谔方程求解后,得到了完全相同的基态电子密度 $n(r)$。
这两个不同的外势对应两个不同的哈密顿量 $\hat{H}_1$ 和 $\hat{H}_2$。 假设它们的基态波函数分别为 $\Psi_1$ 和 $\Psi_2$(这里先讨论非简并基态),对应的基态能量为 $E_1$ 和 $E_2$。 根据薛定谔方程:
\[\hat{H}_1 \Psi_1 = E_1 \Psi_1\] \[\hat{H}_2 \Psi_2 = E_2 \Psi_2\]由于 $\Psi_1$ 是 $\hat{H}_1$ 的唯一基态波函数,任何其他波函数在 $\hat{H}_1$ 下的能量期望值都必然严格大于 $E_1$。我们将 $\Psi_2$ 作为试探波函数代入 $\hat{H}_1$,理应有
\[E_1 < \langle \Psi_2 | \hat{H}_1 | \Psi_2 \rangle\]由于 $\hat{H}1 = \hat{H}_2 + \hat{V}{ext}^{(1)} - \hat{V}_{ext}^{(2)}$,我们将上式展开:
\[E_1 < \langle \Psi_2 | \hat{H}_2 + \hat{V}_{ext}^{(1)} - \hat{V}_{ext}^{(2)} | \Psi_2 \rangle\] \[E_1 < \langle \Psi_2 | \hat{H}_2 | \Psi_2 \rangle + \langle \Psi_2 | \hat{V}_{ext}^{(1)} - \hat{V}_{ext}^{(2)} | \Psi_2 \rangle\]| 因为 $\langle \Psi_2 | \hat{H}_2 | \Psi_2 \rangle = E_2$,且根据算符定义,外势能期望值可以用电子密度表示为 $\int n_2(r) V_{ext}(r) dr$。于是上式变为: |
同样的逻辑可以把符号调转一下得到
\[E_2 < E_1 + \int n_1(r) \left[ V_{ext}^{(2)}(r) - V_{ext}^{(1)}(r) \right] dr\]由于我们假设这两个系统的基态电子密度是相同的,即 $n_1(r) = n_2(r) = n(r)$。将这个条件代入上述两个不等式
\[E_1 < E_2 + \int n(r) \left[ V_{ext}^{(1)}(r) - V_{ext}^{(2)}(r) \right] dr\] \[E_2 < E_1 + \int n(r) \left[ V_{ext}^{(2)}(r) - V_{ext}^{(1)}(r) \right] dr\]两式左右分别相加
\[E_1 + E_2 < E_1 + E_2\]因此直接得到初始的假设错误。结论成立:基态电子密度 $n(r)$ 与外势 $V_{ext}(r)$ 之间存在一一对应的映射关系。 而唯一能够区分不同物理系统(比如水分子 vs. 铁块)的,其实也只有外势 $V_{ext}(r)$。
由于外势唯一决定了哈密顿量[3],而哈密顿量决定了系统的所有性质,因此可以大胆说系统的各种基态性质都可以写成基态电子密度 $n(r)$ 的唯一泛函。例如对于能量
\[E[n] = T[n] + V_{ee}[n] + \int n(r)V_{ext}[n]dr\]其中 $T[n]$ 是电子动能泛函,$V_{ee}[n]$ 是电子间的相互作用能泛函。
可喜可贺可喜可贺。但这个定理并没有告诉我们,如果给定外势 $V_{ext}(r)$,其对应的具体的电荷密度应该如何求出,为此还需要稍微更进一步。
1.3 HK 第二定理(变分原理)
Theorem 2:对于给定的外势 $V_{ext}(r)$,可以定义一个关于电子密度 $n(r)$ 的全能泛函 $E_v[n]$。当且仅当输入真实的基态电子密度 $n_0(r)$ 时,该泛函取得全局最小值,且极小值等于系统的真实基态能量 $E_0$。
根据Theorem 1,由于 $n(r)$ 唯一定义了系统的哈密顿量,它自然也唯一对应着基态波函数 $\Psi$。前面已经提到,系统的总能量泛函可以写为:
\[E[n] = T[n] + V_{ee}[n] + \int n(r)V_{ext}(r)dr\]由于只有基态波函数和基态电荷密度有这种一一对应关系。现在,如果对于一个确定的外势 $V_{ext}(r)$,我们输入一个错误的、非基态的试探电子密度 $n’(r)$。根据第一定理,这个 $n’(r)$ 必然对应另一个不同的波函数 $\Psi’ = \Psi[n’]$。 我们根据量子力学的传统变分原理,用错误的波函数 $\Psi’$ 去计算正确哈密顿量 $\hat{H}$ 的能量期望值,结果必然大于基态能量 $E_0$:
\[E_0 < \langle \Psi' | \hat{H} | \Psi' \rangle\]也就是说,真实的基态电子密度 $n_0(r)$ 能够使能量泛函最小化。在实际计算中,只要对 $E[n]$ 进行泛函求导(满足电子数守恒条件 $\int n(r)dr = N$),即可解出真实的基态密度。[4]
2. KS方程与交换相关能拟设
2.1 Kohn-Sham方程
在HK定理的讨论中,我们似乎通过一一对应的方式很自然地用起了动能 $\hat{T}$ 和电子排斥 $\hat{V}_{ee}$的泛函,但实际上我们还是没法显式地写出它们关于电荷密度$n$的直接表达式,依然只能用最朴素的定义
\[T[n] = -\frac{1}{2} \sum_{i=1}^N \int \Psi^*(x) \nabla_i^2 \Psi(x) dx\] \[V_{ee}[n] = \sum_{i < j}^{N} \int \Psi^*(x) \frac{1}{|\mathbf{r}_i - \mathbf{r}_j|} \Psi(x) dx\]这肯定是没办法让人接受的,为了对方程做简化,我们尝试虚构一个完全没有相互作用的单粒子参考系统:
-
假设该系统包含 $N$ 个没有相互作用的非真实电子。
-
因为没有相互作用,该系统的全同多体波函数可以精确地写为一个斯莱特行列式 $\Phi_{KS}$,它由一组单粒子波函数 $\psi_i(r)$构成。[5]
-
为了与真实体系对应,强加一个虚拟的有效势场 $V_{eff}(r)$,使得这个虚拟系统的基态电子密度,恰好等于真实体系的真实基态电子密度。
在这个体系下,电子密度可以由单粒子轨道直接模平方求和得到
\[n(r) = \sum_{i=1}^N |\psi_i(r)|^2\]同时,单粒子轨道相当于系统的本征态,满足正交条件。
\[\int \psi_i^*(r) \psi_j(r) dr = \delta_{ij}\]那么现在我们可以自然地写出这个系统的动能$T_s[n]$和电子相互能$E_H[n]$
\[T_s[n] = -\frac{1}{2} \sum_{i=1}^N \langle \psi_i | \nabla^2 | \psi_i \rangle = -\frac{1}{2} \sum_{i=1}^N \int \psi_i^*(r) \nabla^2 \psi_i(r) dr\] \[E_H[n] = \frac{1}{2} \iint \frac{n(r)n(r')}{|r - r'|} drdr'\]这两项肯定会和真实系统有所差异,但我们可以把真实系统里所有“算不清、写不出纯密度公式”的尾巴强行扔进一个叫交换相关能(Exchange-Correlation Energy)的垃圾桶里,这也是整个DFT理论里最dirty的地方
\[E_{xc}[n] \equiv (T[n] - T_s[n]) + (V_{ee}[n] - E_H[n])\]至此,真实系统的总能量泛函被改写为:\(E[n] = T_s[n] + E_H[n] + \int n(r)V_{ext}(r)dr + E_{xc}[n]\)
接下来,为了寻找让总能量最低的真实基态密度,我们需要对上述能量泛函进行变分。但由于 $T_s[n]$ 是通过轨道 $\psi_i(r)$ 表达的,我们需要直接对单粒子波函数 $\psi_i^*(r)$ 进行变分。
这部分推导有点烦人,skip
总之通过一通操作我们可以得到下面的单粒子轨道方程
\[\left[ -\frac{1}{2}\nabla^2 + V_{ext}(r) + V_H(r) + V_{xc}(r) \right] \psi_i(r) = \epsilon_i \psi_i(r)\]其中
-
外部势场 $V_{ext}(r) = -\sum_{I=1}^M \frac{Z_I}{ r - R_I }$ -
Hartree势 $V_H(r) = \int \frac{n(r’)}{ r - r’ } dr’$ - $V_{xc}(r) = \frac{\delta E_{xc}[n]}{\delta n(r)} = 不知道什么玩意$
不妨定义有效势场 $V_{eff}(r)$
\[V_{eff}(r) = V_{ext}(r) + V_H(r) + V_{xc}(r)\]于是我们便得到了大名鼎鼎的 Kohn-Sham 单粒子方程 [6]
\[\boxed{ \left[ -\frac{1}{2}\nabla^2 + V_{eff}(r) \right] \psi_i(r) = \epsilon_i \psi_i(r) }\]KS方程在1965年提出,是的没错,Kohn的两篇重量级论文之间只间隔了一年。
求解这些单粒子方程后,我们就可以近似得到整个真实体系的波函数。可喜可贺。
2.2 交换相关势
wait,我们显然不能把$V_{xc}(r)$视而不见,它直接来自我们凑出来的交换相关能$E_{xc}$。但由于$E_{xc}$包含了多个复杂效应(主要是交换能和相关能),导致甚至很难定性地给它一个描述。
但是没办法,这部分能量不能直接无视,只能硬着头皮去凑一个泛函出来。
在最经典的局域密度近似 LDA 中,我们假设,真实体系(如分子、晶体)中某一点 $r$ 的交换相关能,恰好等于一个拥有“相同电子密度”的均匀电子气在该密度下的交换相关能。
基于这个粗糙的假设,真实体系的 LDA 交换相关能泛函可以写成空间积分的形式
\[E_{xc}^{LDA}[n] = \int n(r) \epsilon_{xc}^{HEG}(n(r)) dr\]$\epsilon_{xc}^{HEG}(n)$为每个电子在密度为 $n$ 的均匀电子气中所拥有的交换相关能。我们可以把交换(Exchange)和相关(Correlation)这两项量子效应拆开分别处理
\[E_{xc}^{LDA}[n] = \int n(r) \left[ \epsilon_x(n) + \epsilon_c(n) \right] dr\]其中的交换能$\epsilon_x(n)$(又称 Dirac 交换能)是有解析形式的
\[\epsilon_x(n) = -\frac{3}{4} \left( \frac{3}{\pi} \right)^{1/3} n^{1/3}\]而$\epsilon_c$只能靠数值拟合,1980年Cepeley用QMC解了均匀电子气的模型后,有人就把论文里离散的数据一通拟合得到了$\epsilon_c(n)$的经验方程。由此DFT才得以真正使用。
接着后人还慢慢发明出GGA、meta-GGA等形式的交换相关能拟设,求解精度确实是在上升,但具体是怎么上升的你就别问了(
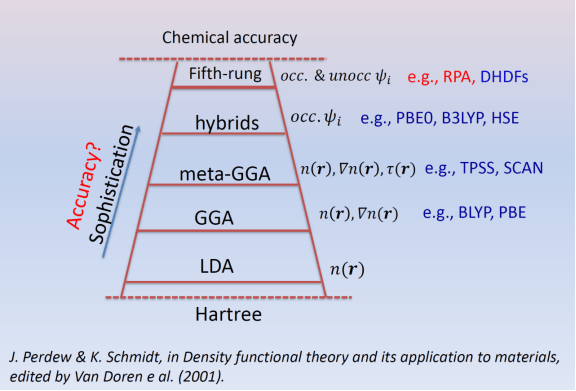
3. 自洽场(SCF)循环
如果你还没有被前面一堆逻辑看上去没什么毛病的式子给糊弄过去,并真正打算动手算算看,你就会发现你还是没法开始。倒不是方程有问题,而是这玩意是耦合在一起的。
-
你想算出虚拟的单粒子波函数 $\psi_i(r)$,给我先算有效势 $V_{eff}(r)$。
-
你想算有效势 $V_{eff}(r)$ ,给我先算电子密度 $n(r)$。
-
你想算电子密度 $n(r)$?很简单,$n = \sum \psi_i ^2$,气笑了。

但说实在这也不是不能做,先猜个解然后慢慢迭代等收敛什么的也就是家常便饭。最不过脑子的设想是下面这样的
Ⅰ.初始化固定项
- 输入原子坐标,生成不变的外势 $V_{ext}(r)$。
- 基组展开:先随便猜几个单电子波函数,一般会用已知基函数(高斯或平面波)的线性组合:$\psi_i = \sum_\mu C_{\mu i} \phi_\mu$。
- 生成初始电子密度$n_{0}(r)$
Ⅱ.开始迭代
- 获取本轮输入密度 $n_{in}$
- 计算有效势 $V_{eff}$
- 计算KS方程,解得新的一组波函数基底${\psi_i}$
- 计算新的电荷密度作为下一轮输入$n_{out}$
大体的思路其实没问题,但是结果会不会收敛是未知数,大概率最后会反复横跳。工业上(数值模拟软件)采用的规范的自洽场求解方式大概是下面这样
一、初始化与基组展开
- 确定系统哈密顿量的固定项:输入原子核坐标 ${\vec{R}_I}$ 和外场,直接构建不变的外势:
- 波函数的基组展开:将未知的虚拟单粒子波函数 $\psi_i(\vec{r})$ 用一组已知的基函数(基础波) $\phi_\mu(\vec{r})$ 进行线性组合展开:
- 计算重叠矩阵 $S$:计算基函数之间的非正交重叠程度:
二、 SCF 迭代
生成初始/当前电子密度 $n^{(k)}(\vec{r})$
- 第 1 步循环 ($k=1$):通过孤立原子电荷密度的线性叠加(SAD)生成一个初始密度 $n^{(1)}(\vec{r})$。
- 后续循环 ($k>1$):使用上一步经由混合算法优化后得到的输入密度 $n_{in}^{(k)}(\vec{r})$。
利用当前的电子密度 $n^{(k)}(\vec{r})$,在空间网格上计算当前轮次的有效势能:
\[V_{eff}^{(k)}(\vec{r}) = V_{ext}(\vec{r}) + \int \frac{n^{(k)}(\vec{r}')}{|\vec{r} - \vec{r}'|} d\vec{r}' + V_{xc}[n^{(k)}(\vec{r})]\]随后,计算哈密顿矩阵的每一个矩阵元(包含动能矩阵 $T$ 和势能矩阵 $V$):
\[H_{\mu\nu}^{(k)} = \int \phi_\mu^*(\vec{r}) \left[ -\frac{1}{2}\nabla^2 + V_{eff}^{(k)}(\vec{r}) \right] \phi_\nu(\vec{r}) d\vec{r}\]KS 方程是偏微分方程,为了数值上能快速求解,将其转化为广义矩阵特征值问题(Roothaan-Hall 方程形式):
\[\mathbf{H}^{(k)} \mathbf{C}^{(k)} = \mathbf{S} \mathbf{C}^{(k)} \mathbf{E}^{(k)}\]求解完成后,挑选出能量最低的 $N$ 个占据态单粒子轨道(就是对应着整个费米子体系的基态),重新组合计算出这一轮对角化自发产生的输出电荷密度:
\[n_{out}^{(k)}(\vec{r}) = \sum_{i=1}^{N_{occ}} f_i \left| \sum_{\mu=1}^{M} C_{\mu i}^{(k)} \phi_\mu(\vec{r}) \right|^2\]同时,根据当前的 $n_{out}^{(k)}$ 算得当前步的系统总能量 $E^{(k)}$。
每次循环后,计算当前步与上一步的残差。只有当能量变化和电荷密度残差同时小于阈值时,循环才允许终止:
-
能量判据:$\Delta E = E^{(k)} - E^{(k-1)} < 10^{-6} \text{ eV}$ -
密度判据:$\Delta n = \int n_{out}^{(k)}(\vec{r}) - n_{in}^{(k)}(\vec{r}) d\vec{r} < 10^{-5}$
如果满足条件就退出循环。但如果不满足条件,不能直接将 $n_{out}^{(k)}$ 代入第 $k+1$ 步。需要调用一些额外的算法生成平滑的 $n_{in}^{(k+1)}$。比如将本轮的输入与输出进行线性加权。
\[n_{in}^{(k+1)} = \alpha n_{out}^{(k)} + (1-\alpha) n_{in}^{(k)}\]又或者可以参考前 $M$ 步(通常 $M=5\sim8$)的历史轨迹。这部分有点类似机器学习的Optimizer,可以有多种选取形式。 [7]
初学者需要了解的内容应该不会超出这些了吧,大概(
那么,以上。
[1] $x_i = (r_i, s_i)$ 是一个复合坐标,包含自旋坐标 $s_i$。
[2] P. Hohenberg and W. Kohn, “Inhomogeneous Electron Gas,” Phys. Rev. 136, B864 (1964).
[3]其实还有一个变量即电子数$N$,但这个同样可由电荷密度直接得到$N = \int n(r) dr$
[4]这里的论证忽略了简并,但我懒得写了,感兴趣的可以查查看,至少核心结论是不会变的。
[5]形式上很像HF方法,但两个模型差异还是很大的。
[7]也由此显得有些不靠谱



